The Computational Biology Platform at CZ Biohub SF develops novel computational methods, models, and tools to advance the Biohub’s research in dynamic cell systems and infectious disease.
Our Team

Led by Yasin Şenbabaoğlu, the Computational Biology Platform collaborates with Biohub SF researchers on the analysis of multi-omic and multimodal datasets as well as development of novel methodologies that advance our understanding of health and disease. In line with CZ Biohub’s mission, we are committed to open science and freely sharing data and code with the broader scientific community.
Our Collaborations and Projects
-
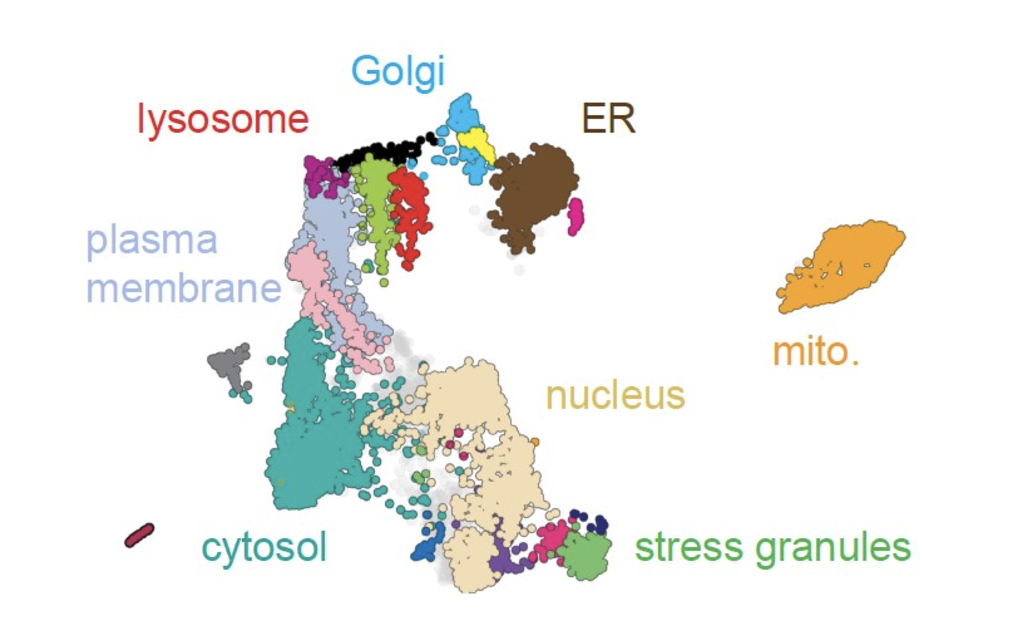 Advanced Organelle Profiling and Subcellular Mapping
Advanced Organelle Profiling and Subcellular MappingWe conduct large-scale studies to profile protein localization across organelles, using an innovative computational approach that enables unprecedented insights into protein dynamics within cells. This groundbreaking technique provides a comprehensive view of subcellular remodeling at the proteome scale in healthy and disease conditions.
-
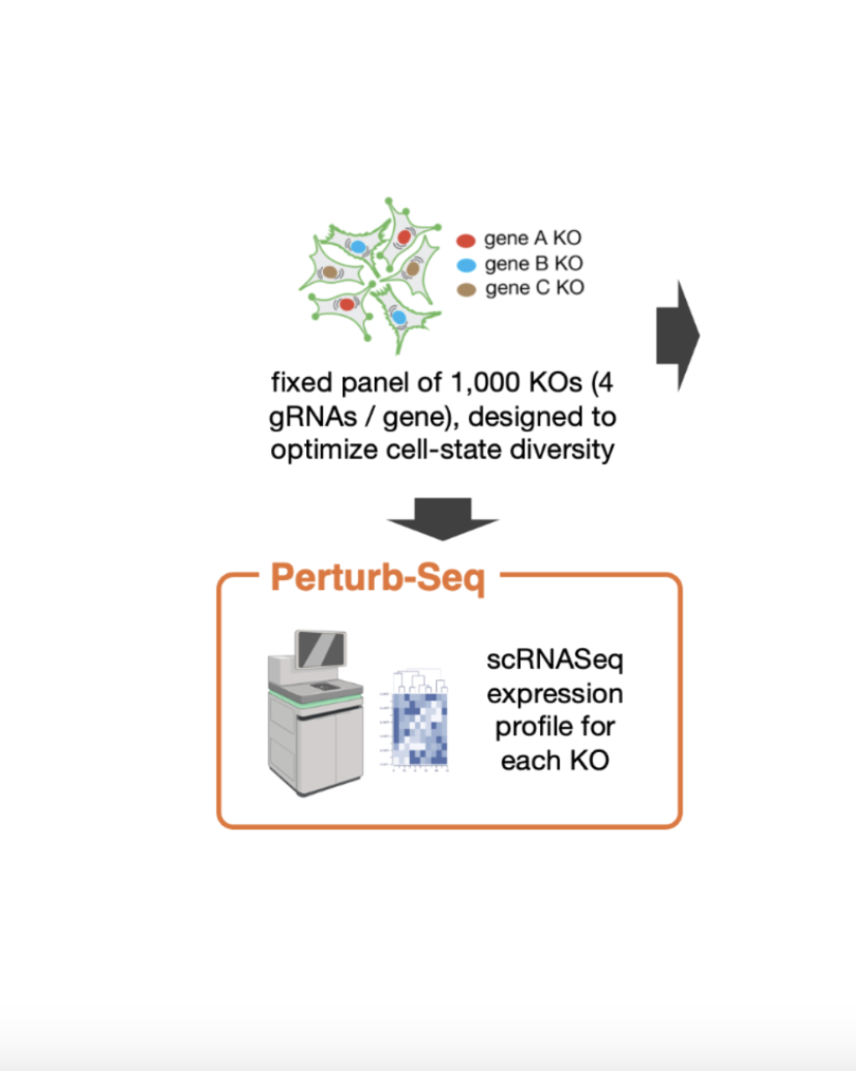 Single-Cell CRISPR Perturbation Screening
Single-Cell CRISPR Perturbation ScreeningOur team designed a 1,000-gene CRISPR panel to systematically explore cellular states and pathways in response to gene knockout. Using both optical pooled screens and single-cell sequencing, we map morphological and transcriptomic changes, targeting critical pathways, such as the unfolded protein stress response.
-
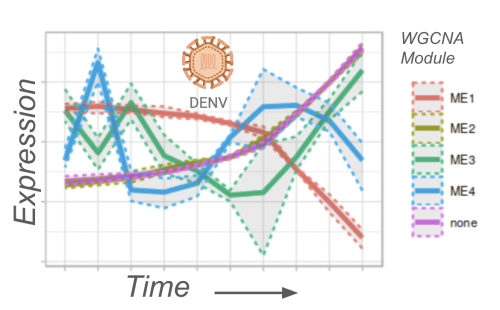 Infectomics and Viral Response Profiling
Infectomics and Viral Response ProfilingWe explore molecular changes in infected cells by profiling gene and protein expression at high temporal resolution, with a focus on Zika and Dengue viruses. This project includes single-cell analyses and imaging to link morphological changes with molecular profiles during infection.
-
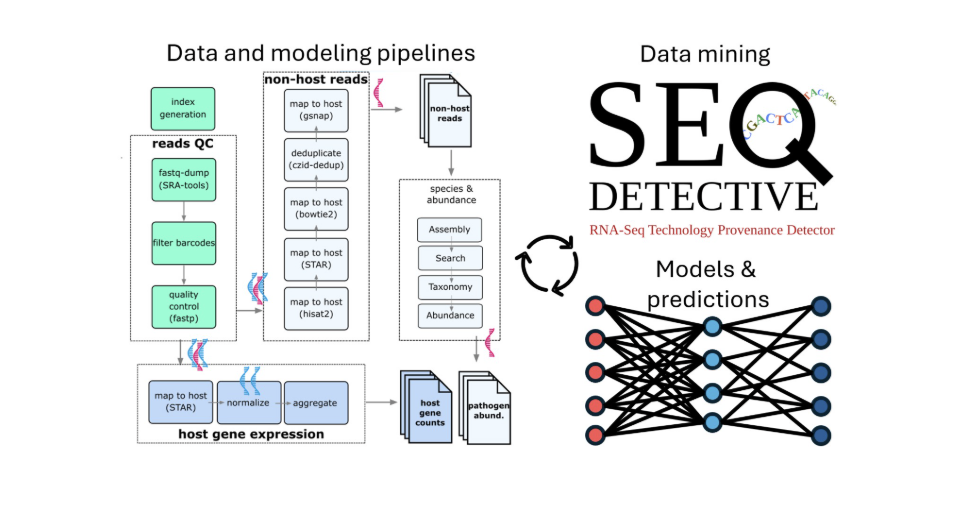
 RNAquarium and Pathogen Discovery Pipelines
RNAquarium and Pathogen Discovery PipelinesBy leveraging public RNAseq data, we identify non-host reads to map potential viruses and pathogens in species like zebrafish. This pipeline is part of our larger effort to create accessible virus-host interaction datasets for broader scientific use.
-
 Building Foundation Models and Machine Learning Pipelines for Model Organism
Building Foundation Models and Machine Learning Pipelines for Model OrganismWe are developing infrastructure and deep learning strategies to standardize data integration across scales and technologies, initially focusing on zebrafish. Using the RNAquarium dataset and other resources, we pretrain and fine-tune models to reveal gene network dynamics in infection, conduct interpretability analyses, and generate hypotheses on host-factor dependencies in pathogens.
-
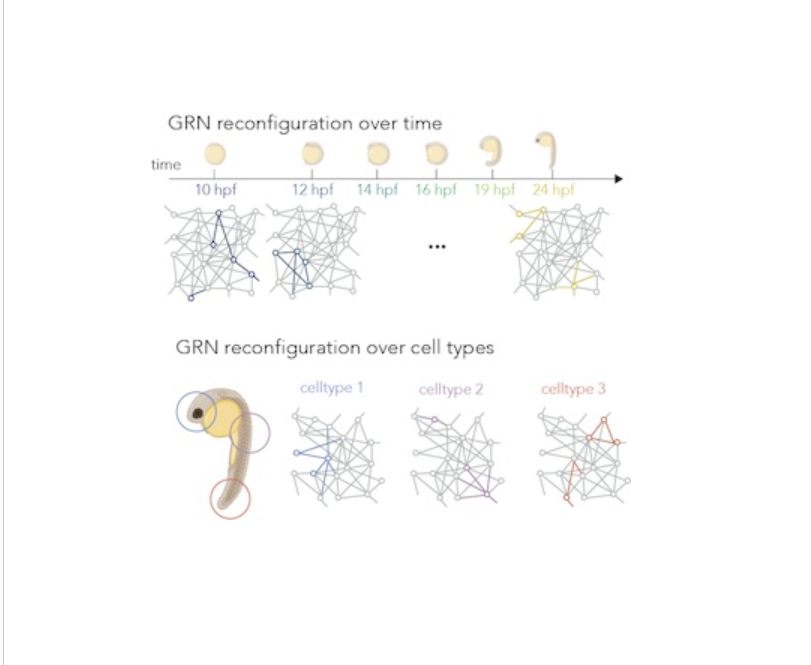 Single-Cell Multi-Omic Atlas Development
Single-Cell Multi-Omic Atlas DevelopmentWe generate comprehensive single-cell multi-omic atlases, such as for zebrafish embryonic development, integrating RNA and ATAC sequencing to map gene regulatory networks across timepoints and cell types. This approach reveals cell-type-specific regulatory modules and enables in silico genetic perturbation studies to explore developmental pathways.
-
 Developing Temporally-Resolved Gene Expression Models for Dynamic Cellular Insights
Developing Temporally-Resolved Gene Expression Models for Dynamic Cellular InsightsLeveraging single-cell multi-omic zebrafish datasets, we are developing temporally-resolved deep learning models to predict cell-type- and condition-specific gene expression dynamics over time. These models will enhance our understanding of regulatory mechanisms as they evolve, with potential applications in tracking cell fates during embryonic development.
-
 Automated Dataset Analysis and Pipeline Optimization
Automated Dataset Analysis and Pipeline OptimizationWe streamline data processing workflows, including mass spectrometry analysis, with a robust sample-to-data pipeline. Currently, we are enhancing these workflows using Nextflow to improve automation, flexibility, and scalability.

