Pete Farley
Director of Communications





Chan Zuckerberg Biohub scientists develop a deep-learning method that reveals rich biological information on proteins and cell architecture.
Aug 2, 2022

AI learned how to recognize and classify different dog breeds from images. A new machine learning method from CZ Biohub now makes it possible to classify and compare different human proteins from fluorescence microscopy images. (Photo collage. Credit: OpenCell/CZ Biohub)
Humans are good at looking at images and finding patterns or making comparisons. Look at a collection of dog photos, for example, and you can sort them by color, by ear size, by face shape, and so on. But could you compare them quantitatively? And perhaps more intriguingly, could a machine extract meaningful information from images that humans can’t?
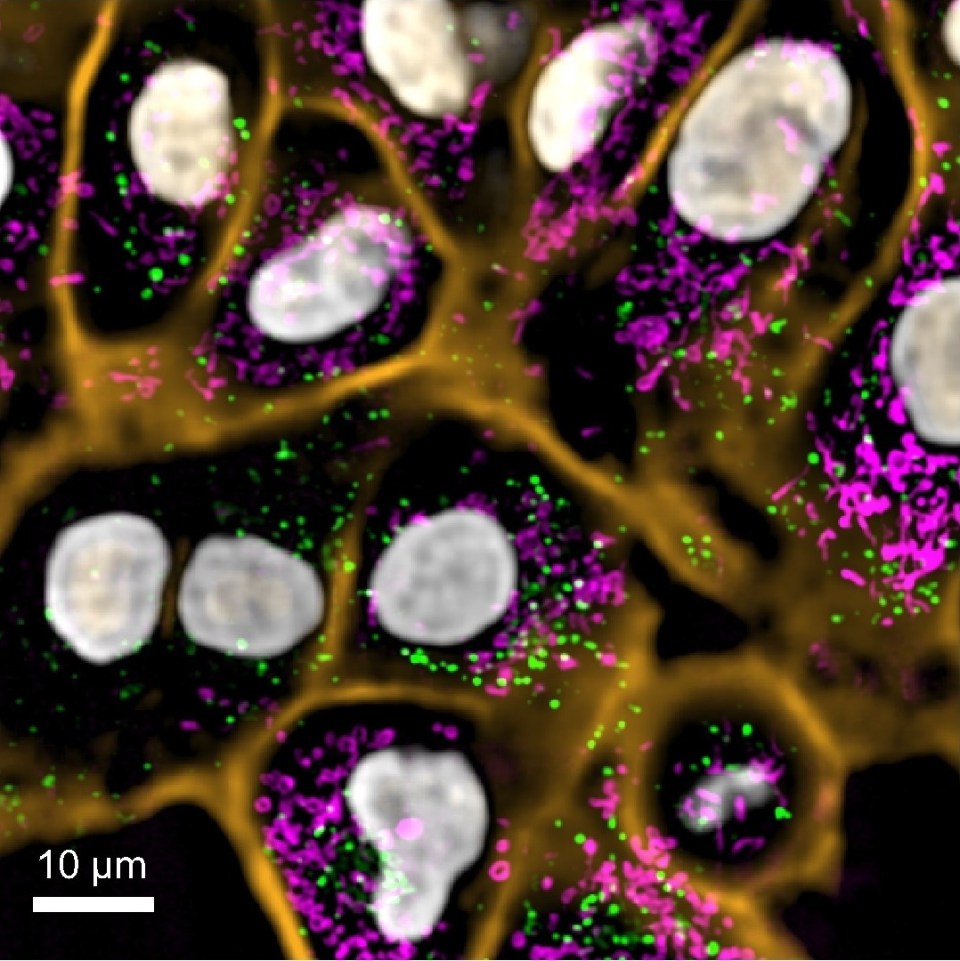
Now a team of Chan Zuckerberg Biohub scientists has developed a machine learning method to quantitatively analyze and compare images – in this case microscopy images of proteins – with no prior knowledge. As reported in Nature Methods, their algorithm, dubbed “cytoself,” provides rich, detailed information on protein location and function within a cell. This capability could quicken research time for cell biologists and eventually be used to accelerate the process of drug discovery and drug screening.
“This is very exciting – we’re applying AI to a new kind of problem and still recovering everything that humans know, plus more,” said Loic Royer, co-corresponding author of the study. “In the future we could do this for different kinds of images. It opens up a lot of possibilities.”
Cytoself not only demonstrates the power of machine-learning algorithms, it has also generated insights into cells, the basic building blocks of life, and into proteins, the molecular building blocks of cells. Each cell contains about 10,000 different types of proteins – some working alone, many working together, doing various jobs in various parts of the cell to keep them healthy. “A cell is way more spatially organized than we thought before. That’s an important biological result about how the human cell is wired,” said Manuel Leonetti, also co-corresponding author of the study.
And like all tools developed at CZ Biohub, cytoself is open source and accessible to all. “We hope it’s going to inspire a lot of people to use similar algorithms to solve their own image analysis problems,” said Leonetti.
Cytoself is an example of what is known as self-supervised learning, meaning that humans do not teach the algorithm anything about the protein images, as is the case in supervised learning. “In supervised learning you have to teach the machine one by one with examples; it’s a lot of work and very tedious,” said Hirofumi Kobayashi, lead author of the study. And if the machine is limited to the categories that humans teach it, it can introduce bias into the system.
“Manu [Leonetti] believed the information was already in the images,” Kobayashi said. “We wanted to see what the machine could figure out on its own.”
Indeed, the team, which also included CZ Biohub Software Engineer Keith Cheveralls, were surprised by just how much information the algorithm was able to extract from the images.
“The degree of detail in protein localization was way higher than we would’ve thought,” said Leonetti, whose group develops tools and technologies for understanding cell architecture. “The machine transforms each protein image into a mathematical vector. So then you can start ranking images that look the same. We realized that by doing that we could predict, with high specificity, proteins that work together in the cell just by comparing their images, which was kind of surprising.”
In this 3D rotation, each point represents a single protein image analyzed by cytoself, colored according to protein localization categories. Collectively, they form a highly detailed map of the full diversity of protein localizations. (Credit: Royer Group / CZ Biohub)
While there has been some previous work on protein images using self-supervised or unsupervised models, never before has self-supervised learning been used so successfully on such a large dataset of over 1 million images covering over 1,300 proteins measured from live human cells, said Kobayashi, an expert in machine learning and high-speed imaging.
The images were a product of CZ Biohub’s OpenCell, a project led by Leonetti to create a complete map of the human cell, including eventually characterizing the 20,000 or so types of proteins that power our cells. Published earlier this year in Science were the first 1,310 proteins they characterized, including images of each protein (produced using a type of fluorescent tag) and mappings of their interactions with one another.
Cytoself was key to OpenCell’s accomplishment (all images available at https://opencell.czbiohub.org/), providing very granular and quantitative information on protein localization.
“The question of what are all the possible ways a protein can localize in a cell – all the places it can be and all the kinds of combinations of places – is fundamental,” said Royer. “Biologists have tried to establish all the possible places it can be, over decades, and all the possible structures within a cell. But that has always been done by humans looking at the data. The question is, how much have human limitations and biases made this process imperfect?”
Royer added: “As we’ve shown, machines can do it better than humans can do. They can find finer categories and see distinctions in the images that are extremely fine.”
The team’s next goal for cytoself is to track how small changes in protein localization can be used to recognize different cellular states, for example, a normal cell versus a cancerous cell. This might hold the key to better understanding of many diseases and facilitate drug discovery.
“Drug screening is basically trial and error,” Kobayashi said. “But with cytoself, this is a big jump because you won’t need to do experiments one-by-one with thousands of proteins. It’s a low-cost method that could increase research speed by a lot.”
Media Contact:
Julie Chao, CZ Biohub
julie.chao@czbiohub.org | 415.816.5571
Stay up-to-date on the latest news, publications, competitions, and stories from CZ Biohub.
Marketing cookies are required to access this form.
For media inquiries, please contact us at media@czbiohub.org

Pete Farley
Director of Communications

Julie Chao
Science Communications Manager
 News Releases
News Releases
Nine researchers receive unrestricted funding for projects to harness the immune system to detect disease and monitor ...
 News Releases
News Releases
The CZ Biohub San Francisco and CZ Imaging Institute will join together to develop novel imaging technologies that provide entirely new insights into human ...
 News Releases
News Releases
The growing team at CZ Biohub NY is shaping the future of life sciences in the New York ...
Stay up-to-date on the latest news, publications, competitions, and stories from CZ Biohub.
Cookies and JavaScript are required to access this form.

